
Malonsäurediethylester (Diethylmalonat) ist in der organischen Synthese einer der wichtigsten und vielseitigsten Synthesebausteine. Mit seinen aciden H lassen sich viele Kondensationsreaktionen ausführen - z.B. sind über Malonestersynthese substituierte Essigsäuren oder Barbitursäurederivate zugänglich, Malonsäureester ist auch Ausgangsbaustein für die Knoevenagel-Reaktion. Der standard-Syntheseweg für Malonsäurediethylester läuft über Chloressigsäure → Cyanessigsäure → Verseifung des Nitrils bzw. Veresterung in alkoholischer Lösung direkt zum Ethylester. Dieser Weg ist aufwändig und erfordert Einsatz von Cyanid bzw. Einleiten von HCl-Gas - nicht gerade das was man unbedingt im Hobbylabor möchte.
Wikipedia sagt: "Malonsäurediethylester kann nicht aus der freien Säure durch Veresterung mit Ethanol gewonnen werden, weil Malonsäure bei erhöhten Temperaturen zu Essigsäure und CO2 zerfällt (Decarboxylierung)". Oder etwa doch? Alles nur eine Frage der richtigen Reaktionsführung! Azeotrope Veresterungen sind ein sehr eleganter Weg das Gleichgewicht gezielt in Richtung Produktseite zu verschieben. Durch geeignete Wahl eines Schleppmittels wird die Reaktionstemperatur niedrig gehalten und Reaktionswasser kontinuierlich entzogen, bis hin zu einer praktisch quantitativen Umsetzung. Für Veresterungen mit Ethanol bietet sich dabei besonders das System mit Chloroform an.
Geräte:
250 ml Rundhalskolben, Wasserabscheider für schwere organische Phasen, Rückflusskühler, Destillationsapparatur für Normaldruck bzw. Vakuumdestillation, Heizplatte mit Magnetrührer (idealerweise mit geregelter Temperatur), Ölbad, Vakuumpumpe mit zwischengeschalteter Sicherheitswaschflasche
Chemikalien:
Malonsäure
Ethanol
Chloroform
p-Toluensulfonsäure

Malonsäurediethylester
Hinweis:
Chloroform ist giftig und wirkt betäubend - unbedingt im Abzug arbeiten!
Bei Vakuumdestillationen besteht immer die Gefahr einer Implosion - als Schutz vor Splittern ist eine Schutzbrille zu tragen!
Durchführung:
Zuerst wurde die Apparatur zur Wasserabscheidung zusammengebaut, dampfführende Rohre wurden gut mit Alufolie isoliert um Wärmeverluste und ungenutzten Rückfluss zu minimieren. Im 250 ml 2-Hals Rundkolben wurden dann 30,0 g (0,288 mol) Malonsäure, 5 g (0,029 mol) p-Toluolsulfonsäure, sowie 60 ml Ethanol (50 % molarer Überschuss bezogen auf die Malonsäure plus Zuschlag für Verluste bei der azeotropen Destillation) und 100 ml Chloroform vorgelegt. Das Heizbad wurde auf 110-120 °C geregelt um einerseits einen kräftigen Rückfluss am Wasserabscheider zu erzielen, andererseits aber keine lokale Überhitzung zu erzeugen, die die Malonsäure zersetzen könnte. Unter kräftigem Rühren wurde nun ca. 2,5 - 3 Stunden lang gekocht, bis sich ca. die abgeschätzte Menge wässriger Phase (ca. 20 ml) gebildet hatte und keine weitere Wasserabscheidung mehr zu beobachten war. Das Kochen wurde beendet, die Reaktionsmischung etwas auskühlen gelassen und die Flüssigkeit aus dem Wasserabscheider vorsichtig abgelassen. Die untere, chloroformreiche Phase wurde aufbewahrt um bei zukünftigen Veresterungen mit Ethanol wieder eingesetzt zu werden. Die obere, wässrige Phase wurde verworfen.
Die Apparatur wurde nun auf eine einfache Destillation mit Liebig-Kühler umgebaut und bei Normaldruck das überschüssige Ethanol und Chloroform abgezogen. Insgesamt destillierten in einem Temperaturbereich von 57 - 61° C noch über 75 ml ab, überwiegend Chloroform bzw. Azeotrop Ethanol-Chloroform. Das Destillat wird aufbewahrt und kann bei zukünftigen Veresterungen mit Ethanol ebenfalls wieder ingesetzt werden. Zusammen mit der Chloroformphase aus dem Wasserabscheider wurden so ca 105 ml wiedergewonnen.
Der Vorstoß wurde nun auf einen Vakuumvorstoß mit Spinne gewechselt. Wiederum wurde zuerst gewartet bis die Reaktionsmischung etwas ausgekühlt war, dann wurde Vakuum angelegt. Nachdem noch die letzten Reste Chloroform abgezogen waren (Vorsicht - der Sumpf kann dabei teils kurz kräftig aufschäumen) wurde das Heizbad wieder aufgedreht und auf ca. 120-130° C geregelt. Nach nur wenigen Tropfen Vorlauf destillierte in einem sehr engen Intervall das reine Produkt in den vorher tarierten Kolben bei 76-77° C über. Bei Anwendung der Antoine-Funktion entspricht das 10-11 mbar. Erfahrungsgemäß erreicht meine Drehschieberpumpe ca. 7-10 mbar (kann ich leider nicht genau messen), was mit dem erwarteten Wert gut übereinstimmt.
Ausbeute: 42,96 g (93,0 % d.Th.)
Entsorgung:
Das wiedergewonnene Chloroform sollte aufbewahrt und für weitere Veresterungen mit Ethanol eingesetzt werden. Sonst zu den halogenhaltigen organischen Abfällen.
Alle anderen Reste kommen zu den organischen Abfällen.
Erklärung:
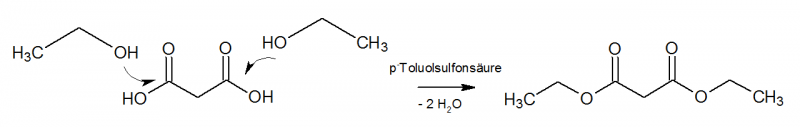
Es erfolgt eine klassische säurekatalysierte Fischer-Veresterung mit Ethanol und Malonsäure
Ein Blick in die "Azeotrope Tables" zeigt folgendes System:
Das ternäre Azeotrop Wasser / Ethanol / Chloroform = 3,5 / 4,0 / 92,0 (alle Angaben in Gewichts-%), siedet bei 55,5° C und zerfällt bei der Kondensation in 2 Phasen. Die schwerere, untere Phase hat die Zusammensetzung (0,5 / 3,7 / 95,8 ), besteht also fast nur aus Chloroform und etwas Alkohol. Die obere hat die Zusammensetzung (80,8 / 18,2 / 1,0), besteht also zum Großteil aus Wasser und nur sehr wenig Chloroform. Das Azeotrop enthält also mit 3,5 % zwar nur relativ wenig Wasser, durch die Phasentrennung kann dieses aber sehr leicht quantitativ abgeschieden werden und man verliert nur wenig Ethanol, das für die Reaktion gebraucht wird, bzw. sehr wenig "Schleppmittel" Chloroform. Die untere, chloroformreiche Phase wird kontinuierlich wieder der Reaktionsmischung zugeführt. Der Siedepunkt ist so niedrig, dass auch empfindlichere Moleküle das gut aushalten. Sofern der gebildete Ester also nicht zu niedrig siedet oder ein anderes, störendes Azeotrop bildet, ist dies ein geradezu ideales System zur schonenden Veresterung.
Weitere Azeotrope bzw. Dampfphasen, die hier mitspielen, sind:
Wasser / Chloroform (2,8 / 97,2) - Siedepunkt 56,1 °C
Ethanol / Chloroform (7,0 / 93,0) - Siedepunkt 59,4 °C
reines Chloroform - Siedepunkt 61,1 °C
Wasser / Ethanol (4,5 / 95,5) - Siedepunkt 78,1 °C
reines Ethanol - Siedepunkt 78,4 °C
reines Wasser - Siedepunkt 100,0 °C
reiner Malonsäurediethylester - Siedepunkt 199 °C
Bilder:

Die fertig aufgebaute Apparatur zur Wasserabscheidung - bereits gut mit Alufolie isoliert

Beginnende Wasserabscheidung - die Chloroformphase ist etwas getrübt da sich durch das Auskühlen die Löslichkeit von Wasser verringert

Die Wasserabscheidung ist beendet

Apparatur umgebaut auf Destillation bei Normaldruck

Vorstoß durch Vakuumvorstoß mit Spinne getauscht

Vakuumdestillation beginnt - immer alles schön kuschelig einpacken! Die Druckanzeige ist nicht wirklich genau, im Wesentlichen nur eine Indikation ob Vakuum anliegt

Das fertige Produkt

...und der verbliebene Rest im Sumpf - zurückgebliebene Toluolsulfonsäure, geringe Restmenge Produkt und diverse Verunreinigungen (selbst das könnte man für eine weitere Veresterung vermutlich wieder einsetzen)


