
m-Methylbenzoesäure ist eine eher unübliche organische Carbonsäure, die sich von der Benzoesäure ableitet. Sie lässt sich durch Amid-Spaltung von N,N-Diethyltoluamid (DEET), einem bekannten Insektenrepellent, herstellen. Obwohl DEET sehr stabil ist, lässt es sich zu einem gewissen Teil in die m-Toluylsäure und das entsprechende Amin spalten.
In dieser Anleitung wird von reinem DEET ausgegangen, das vorher aus Insektenspray isoliert wurde. Im Prinzip bietet aber die ursprüngliche Zusammensetzung des Sprays schon gute Bedingungen für die hier vorgenommene Spaltung, weswegen eine vorangehende Extration des Reinstoffs in diesem Zusammenhang nicht nötig ist.
Geräte:
500 mL-Rundkolben, Bechergläser, Rückflusskühler, Destillationsapparatur- und Zubehör, Filterzubehör, Scheidetrichter, Trocknungsmöglichkeit, Magnetheizrührer, Sandbad.
Chemikalien:
N,N-Diethyltoluamid

Natriumhydroxid

Methanol oder Ethanol


Salzsäure 37%
m-Methylbenzoesäure

Hinweis:Methanol ist giftig, weswegen eher auf Ethanol zurückgegriffen werden sollte. Es wird mit heißer Natronlauge gearbeitet, weswegen auf jeden Fall Schutzkleidung getragen werden sollte.
Durchführung:
9,5 g (0,05 mol) DEET werden in einem Becherglas in 25 mL Methanol gelöst und diese Mischung in einen 500 mL Zweihalskolben überführt, welcher im Sandbad auf einem Heizrührer unter Rühren erhitzt wird. Dabei wird zunächst nur leicht die Temperatur erhöht. Seperat werden 10,0 g (0,25 mol) Natriumhydroxid in 25 mL dest. Wasser gelöst (Hitzeentwicklung!). Die so erhaltene Natronlauge wird zur methanolischen DEET-Lösung gegeben und der Reaktionsansatz mit einem Thermometer versehen. Es wird über 13 Stunden hinweg am Rückfluss gekocht (T = ca. 70-75°C).
Danach tauscht man den Rückflusskühler gegen eine Destillationsbrücke aus und entfernt das Methanol, soweit es geht. Hiernach lässt man abkühlen und trennt die Mischung nach Zugabe von etwas dest. Wasser im Scheidetrichter auf, wobei die wässrige untere Phase weiterverarbeitet wird, indem sie unter Rühren mit 25 mL konz. HCl neutralisiert wird. Dabei ist bereits eine leichte Trübung wahrzunehmen. Beim Abkühlen auf Raumtemperatur scheiden sich fluffige Kristallnadeln ab, welche abfiltriert, in heißem Wasser umkristallisiert und getrocknet werden. Der Schmelzpunkt des Produkts lag bei ca. 115°C (Literatur: 111°C).
Ausbeute: 190 mg (2,75 % d.Th.) eines faserigen, weißen Pulvers ohne nennenswerten Eigengeruch.
Die erhaltene Ausbeute ist sehr, sehr schlecht. Beim Auftrennen der Phasen fiel auf, dass sogut wie alles an DEET noch vorhanden war. Es ist also noch viel Spielraum nach oben. Zur Erhöhung der Effizienz könnten z.B. höhersiedende Alkohole als Lösungsmittel verwendet werden (1-Propanol), allerdings ist es fraglich, ob man generell über diesen Weg der Amidspaltung jemals "gute" Ausbeuten bekommen wird.
Entsorgung:
Die organische Phase besteht quasi nur aus übriggebliebenen DEET und kann für andere Experimente oder eine Wiederholung dieses verwendet werden. Die wässrigen Lösungen werden neutralisiert und im Abfluss oder anorganischem Müll entsorgt.
Erklärung:
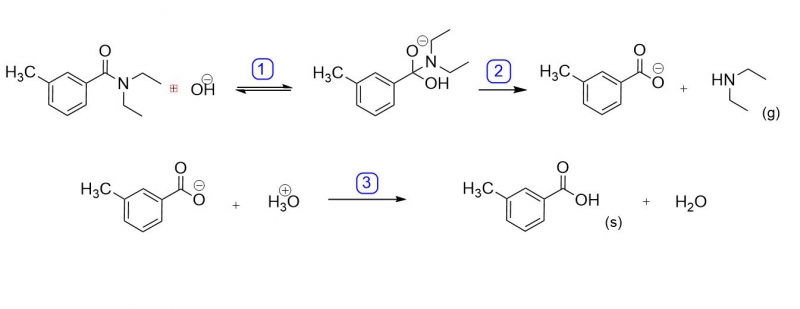
DEET ist ein arom. Carbonsäureamid. Obwohl Amide zu den reaktionsträgsten und damit auch stabilsten Carbonylverbindungen gehören, lassen sie sich durch langes Kochen im Sauren oder Alkalischen spalten. Die Spaltung wird hier im basischen unter NaOH-Zugabe vorgenommen.
Zunächst gehen sowohl das DEET als auch das NaOH in Lösung und können miteinander reagieren, was weder in reinem Wasser noch MeOH/EtOH entsprechend funktionieren würde. Das Hydroxid-Anion greift nukleophil an das Carbonyl des DEET an (1), es entsteht ein tetraedisches Zwischenprodukt. Unter Abspaltung der Amingruppe und weiterer Deprotonierung bildet sich schließlich das Anion der Carbonsäure (2).
Wird die wässrige Phase, welche das m-Methylbenzoat enthält, mit Salzsäure versetzt, entsteht wieder die freie Säure (3), welche in Wasser eine sehr schlechte Löslichkeit von ca. 1g/L besitzt. Sinkt die Temperatur der neutralisierten Lösung (welche zunächst durch Reaktionswärme noch heiß ist) beginnt sich die m-Methylbenzoesäure in wohlgeformten Kristallen abzuscheiden.
Da die Löslichkeit des Produkts in heißem Wasser deutlich höher ist als in kaltem, ist eine Reinigung durch Umkristallisieren in Wasser möglich, was sich bei der extrem niedrigen Ausbeute hier aber nicht lohnte.
Bilder:

Rückflusskochen der Mischung

Abtrennen der wässrigen Phase

Ansäuern der wässrigen Phase

Beim Abkühlen ausfallende Kristallnadeln

Getrocknete m-Methylbenzoesäure

